(Disclaimer: The following experiments do not constitute rigorous peer review, but rather illustrate typical yields obtained and observations gleaned by trained synthetic chemists attempting to reproduce literature procedures. We've taken efforts to stay close to the original procedure, using similar glassware, equipment, and reagents wherever possible. Images have been cropped and scaled to fit in the allotted space, but have not been digitally altered otherwise.)
Updated: 2/20/13 - Added Yu rxn pictures
Ref: Ye, M.; Gao, G.-L.; Yu, J.-Q.
J. Am. Chem. Soc. 2011,
133, 6964-6967. doi:
10.1021/ja2021075.
Recommendation: Pending Blog Syn repeat
Initial - Moderately reproducible - yields/conversion lower than reported; reactivity / regioselectivity mostly consistent with literature. May require optimization.
General scheme:

Context: This reaction comes from a 2011 paper (
dx.doi.org/10.1021/ja2021075) by
Jin-Quan Yu's group at the Scripps Research Institute titled "
Ligand-Promoted C-3 Selective C- H Olefination of Pyridines with Pd Catalysts."
See Arr Oh proposed this reaction, noting its potential to be "operationally simple...with few variables to goof up, and wide substrate availability." Synthetically,
C-H activation appeals to synthetic chemists due to its high atom efficiency (less waste) and simplicity (no need to pre-functionalize a reaction center).
The procedure employs a Pd catalyst to 'activate' the pyridyl C-3 carbon-hydrogen bond. A bidentate pyridyl ligand,
phenanthroline (phen), presumably enhances ligand exchange via the
trans-effect (to counteract the strong coordination of the pyridine substrate to the metal center). The reaction is run with a Ag(I) co-catalyst under air, which serves as the terminal oxidant.
The authors highlight the important of 3-alkenyl pyridine derivatives in chemistry, citing numerous natural products and pharmaceuticals exhibiting this motif. They claim the first instance of C-3 selective pyridine olefination (a number of examples with more limited substrate scope and different site selectivity are given).
Just how C-3 selective? Yu and coworkers report C-3/C-2/C-4 site selectivity ranging from 7/1/1 to 30/1/1.
According to Web of Science, the article has been cited 45 times as of February 2013 (including 4 reviews). A cursory SciFinder search didn't find any examples with direct usage of this chemistry; an extension to arylation is found in a 2011
synthesis of preclamol by Yu's group.
Update(2-17): A related reaction for C-2 olefination of pyridines was reported in Adv. Synth. Cat. 2012,
354, 2135.
We apologize for this omission.
Experimental
Trial 1 - See Arr Oh
Scale: 0.5 mmol (same as researchers)
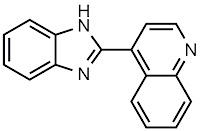
Entry: Compound
3a, Table 2 from
JACS 2011, 6964.
 |
Reaction in 75 mL
sealed pressure tube. |
Reagents: ACROS Ethyl acrylate [
140-88-5], 99.5%; Alfa silver carbonate [
534-16-7], 99.5%; ALD anhydrous pyridine [
110-86-1], 99.8%; STREM Pd(OAc)
2 [
3375-31-3], 99%; ALD 1,10-phen [
66-71-7], Aldrich DMF, anh. 99.8%
Glassware: Pressure tube (75 mL, Teflon bushing) oven-dried o/n. All dry reagents weighed out in air.
Observations: First 5 mins: yellow pasty suspension. At temp (141.3 deg, sand bath) - deep orange-brown. After 16 h - deep brown, almost homogeneous, silvery Ag(0) / Pd(0) sheen around top of flask. Diluted with EtOAc, filtered over 1/2" Celite plug (2 x 5mL wash), then column chromatography (3 x 1 cm, silica, 1:4 - 1:1 EA / Hept).
Pale yellow oil, 7/1/1
meta / ortho / para, 27% yield overall (38 mg isol'd: 24 mg pdt + 14 mg DMF by NMR integration)
Advice: Crude product an orange gum, tough to handle / redissolve. I would re-load onto silica, and incorporate a water wash to eliminate DMF.
Supplementary images (click to enlarge):
 |
| 1H NMR spectrum, post-chromatography. |
 |
| Reaction setup, before heating. |
 |
TLC: left, ethyl acrylate
center, co-spot
right, reaction mixture |
 |
| Filter. |
 |
| Reagents employed. |
 |
| Measured reagents. |
Trials 2 and 3 - Organometallica
Scale: 0.5 mmol, as per SI conditions.
Entry: Compound
3a, Table 2 from
JACS 2011, 6964.
Reagents: Palladium acetate [
3375-31-3] (Strem, fresh bottle stored in glovebox), 1,10-phenanthroline [
66-71-7] (Sigma-Aldrich, age unknown), Silver Carbonate [
534-16-7] (Strem, ~2 mos old, stored in freezer in amber bottle within glovebox), ethyl acrylate [
140-88-5] (Sigma, age unknown-purity >99% by NMR), Pyridine [
110-86-1] (ACROS 99.8%), and DMF [
68-12-2] (from solvent still, source unknown, see
Advice for comments on this reagent).
Glassware: Pressure tube (75 mL).
 |
| Trial 3; reaction setup. |
Observations: NOTE: This reaction was attempted multiple times. In all cases, the reaction was set up on the bench without precautions against preventing air contamination. Reagents were added neat to 75 mL pressure vessel in the order Pd(OAc)
2, 1,10-phenanthroline, Ag
2CO
3, pyridine, ethyl acrylate, followed by DMF, then sealed and heated at 140˚C for 12 hrs.
For
Trial 2, DMF used directly from solvent still, for
Trial 3, air was bubbled through DMF for 1 hour prior to use.
Trial 2: Reaction darkens, remains heterogeneous.
Trial 3 (Pictured): Turns pasty yellow, still some solid left in the flask.
Analysis:
1H NMR:
Trial 2: no conversion observed.
Trial 3: ~40% conversion.
Advice: When I first ran this reaction (
Trial 2), I panicked. There was absolutely no product; nothing by TLC, nothing by NMR, nothing by GC-MS. However, one of my friends who work on Pd-catalyzed C-H activation remarked that reactions run under air are particularly sensitive to solvent O
2 concentration, so much so that he saturates solvents with O
2 before use. Thinking this might be what plagued me, I aerated my solvent (
Trial 3), which seemed to kick off the reaction. This may be the first time I've ever had a reaction fail for having my reagents be
too clean!
Supplementary images (click to enlarge):
 |
| 1H NMR spectrum, full |
 |
1H NMR spectrum, expanded to show
aromatic region |
Results and Discussion
Author* response: Corresponding author
Jin-Quan Yu (Scripps) was kind enough to respond with some helpful advice (
Bold highlights are mine).
1. As what you have observed, the oxygen concentration is very important to this reaction. So the tube volume is also very important. We used the 50-mL tube, which is the total volume of the tube (from the bottom to the cap). For smaller one, the air is not enough. For bigger one, the pyridine will volatilize too much outside the solution.
2. Heating and stirring is another important thing. Prior to starting experiments, turn on the hot plate and set the oil bath to 140°C first. To make the oil temperature is stable, you can put a paper clip into the oil, which will help to stir the oil. Put the reaction tube in the middle of hot plate to obtain a stable stirring (500 rpm is enough). Don't let the solid adhere to the tube wall. And make sure the surface of reaction solution is lower than oil surface.
3. Another thing I should mention is that the quality of Ag2CO3 and Phen may ruin this reaction. We found that these chemicals from Strem Chemicals are good quality and there is no risk for this reaction.
Roundup: The reaction certainly works, though certain variables essential to its success are under-emphasized in the
original paper (see
Author response) and were unfortunately omitted from the
SI. Notably, conversion depends on the presence of oxygen in solution (see Organometallica's trials). Even solvent aeration produced substantially lower conversion than the authors report (40%
vs. 73% isolated yield). See Arr Oh did, however, observe similar regioselectivity (7/1/1
vs. 12/1/1).
Since both yields and observed regioselectivities fall within the low end of those reported, we classify this procedure as
moderately reproducible.
Thanks to See Arr Oh and Organometallica for experiments, and thanks to Prof. Yu for providing feedback. Readers: if you have experience with this reaction or suggestions for future investigations, please feel free to comment.
*Addendum (Feb 20, 2013): After an exchange of emails, Prof. Yu felt it best to have a new postdoc (not an original paper author) repeat the experiment, and sent over pictures of the reaction setup, along with a crude NMR.
The ratio I get from these integrations is 12.7 / 1.5 / 1 (
m/o/p), which agrees with the original publication.
Update (2/20/13): Prof. Yu indicates that integration of the crude against 0.5 equiv. of CH2Br2 (4.94 ppm, above) gives an NMR yield of meta-olefination product matching the literature (76%).
Of note, the reagents are quite different - he's using a higher grade of both (1,10-phen) and pyridine, but we don't have information on the acrylate used or the Pd source. Also of note, the 50 mL long, cylindrical tube volume clearly benefits the reaction, as alluded to in Prof. Yu's original email.
We thank Prof. Yu for sending along some clarification. Perhaps this signifies a change in the way reactions are reported; if
everyone had to send along photo evidence, would there be so many reproducibility issues in the modern literature?
-
SAO